Introduction
As a medical device Notified Body (SGS Belgium NV – Notified Body 1639), we are accredited to certify all types of medical devices, including those without an intended medical purpose. Once certified with us, you will be entitled to use the CE 1639 mark on the devices and their labeling before placing the devices on the European Union market.
All devices certified by SGS NB 1639 require you to get an EU Quality Management System (QMS) certificate. Before using the CE 1639 mark, Class III and implantable Class IIb1 must also have an EU Technical Documentation Assessment certificate.
A list of standard fees for conformity assessment activities under MDR (2017/745) is available from our EU Medical Devices Regulations Information Center.
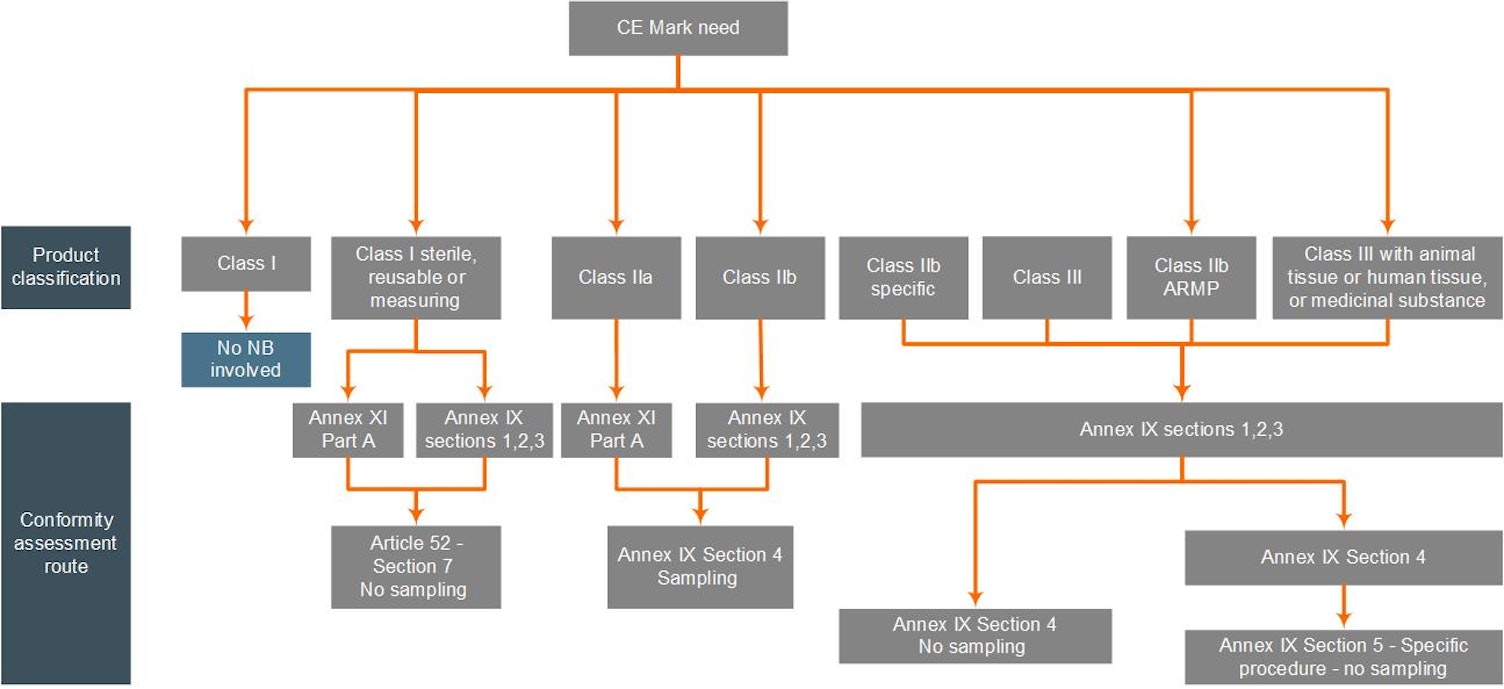
The first step will be for you to determine your product(s) classification according to the rules defined in Annex VIII of the MDR. Subsequently, you must decide which type of conformity assessment path you wish to apply, which is either:
- Conformity assessment based on the QMS and Technical Documentation Assessment as per Annex IX of the MDR
- Conformity assessment based on Product Conformity Verification (Product Quality Assurance) as per Annex XI Part A of the MDR
For devices that are “medicinal” and governed by Directive 2001/83/EC, according to Article 8 of the MDR, we can propose an assessment, according to MDR Article 117, to provide a Notified Body opinion on compliance with GSPR for the medical device part of a drug-device combination. We can also provide QMS certificates to distributors or importers carrying out any activities mentioned in points (a) and (b) of MDR Article 16(2), subject to an application and audit procedure. The sections applicable to these types of assessments will be indicated in this document.
The diagrams below present the type of conformity assessment per device class and guide you through the appropriate certification process that we may offer. For this diagram:
- “Class I Reusable” is an abbreviation of “Class I reusable surgical instruments”
- "Class IIb Specific” are implantable Class IIb devices, except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling
On this page
- Important Information
- Lodging Your Application
- Pre-Application Review
- Contract Proposal
- Application Review
- Stage 2 Audit
- Technical Documentation Assessment
- Nonconformance and Corrective Actions Request
- Certification Review
- Unannounced Audit After CE Certification
- Recertification
- Notification of Changes
- Vigilance
- Summary of Safety and Clinical Performance (SSCP)
- Periodic Safety Update Report (PSUR)
- Voluntary Change of Notified Body
- Additional SGS Medical Device Certification Services
- Useful References
- Annex 1: Changes That Must Be Notified to SGS Before Implementation
- Annex 2: Corrective Action Request (CAR)
Overall conformity assessment process for QMS Audit and Technical Documentation Assessment
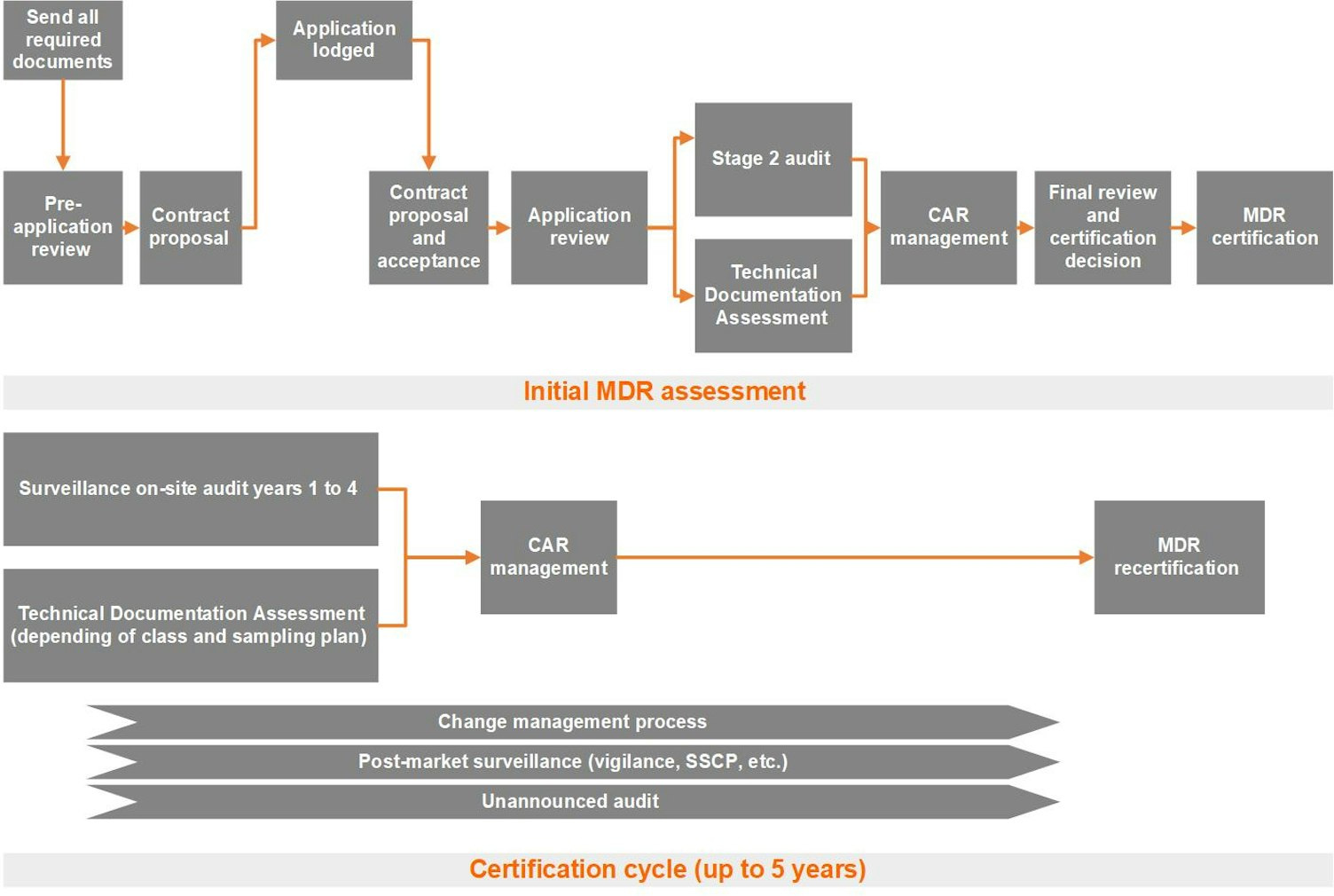
The certification process we may offer
Important information
The certification cycle is normally five years. However, we may shorten the cycle to four years or less, based on the results of the initial, surveillance or recertification conformity assessment. We may also shorten the cycle due to other factors, such as vigilance issues or unannounced audit findings, as stipulated in the MDR. Throughout the certification cycle, we will periodically, at least once every 12 months, carry out surveillance audits and assessments to ensure that your approved quality system remains effective and that certified products remain safe and perform as intended.
Your application and subsequent correspondence must be in English. We can generally accept that your QMS is in your local language or English. Your technical documentation and any further evidence in response to corrective action requests should be submitted in English.
After you receive your CE certification and, in the event of any developments that will alter the scope of your current certification (change of site or product range, reductions in scope, change in company name, etc.), you must inform us as soon as possible and always before implementing the change. You must wait for our approval to implement the change. Please consult Annex 1 of this document for a list of changes for which prior notification is required.
Please note that the conditions set out in the SGS Codes of Practice, General Conditions for Certification and Regulations Governing the Use of SGS Certification Marks apply and can be downloaded from the SGS website.
Do not hesitate to contact your SGS Delivering Office for more information about the certification process.
Lodging your application (sending all required documents)
To apply for certification and start the assessment process, you need to complete, sign and submit both the Medical Device Questionnaire and the Product Information Questionnaire.2 These forms can be downloaded from our EU Medical Devices Regulations Information Center page and should be sent to your local SGS Delivering Office.
In the Medical Device Questionnaire and Product Information Questionnaire(s), the following must be clearly indicated:
- If your devices contain a component/element:
- That has or may have a possible pharmacological, immunological, metabolic or antimicrobial activity (according to the European Medical Device Regulation (hereafter MDR) 2017/745
- That contains animal tissue or derivatives thereof (according to Regulation (EU) No 722/2012)
- That contains phthalate (according to MDR (EU) 2017/745 Annex I Section 10.4.3)
- That contains human cells, blood, tissue or derivatives thereof (according to European Directive 2004/23/EC)
- Substance(s) that is partially or fully absorbed, or undergoes a chemical change in the body
- That contains nanomaterials (e.g. nano-hydroxyapatite, nano-silver) or may generate nanosized particles (e.g. due to wear-and-tear) according to Commission Recommendation 2011/66/EU of October 18, 2011
- If any critical processes are subcontracted or outsourced, copies of any relevant subcontractor/supplier certification should be sent with the Relevant Subcontractor and Supplier list, available via the SGS medical device website
By submitting the Medical Device Questionnaire, as well as the Product Information Questionnaire(s), you confirm that as the legal manufacturer of the device you:
- Have an up-to-date documented quality system available for audit by SGS
- Fulfill the obligations imposed by the quality system
- Have a description of the procedures in place to ensure that the QMS remains adequate and effective, and the undertaking by the manufacturer to apply those procedures
- Did not lodge an application with another Notified Body for the same product-related quality system
- Have up-to-date technical documentation available for assessment by SGS NB 1639. This must contain or refer to documents that contain:
- All the QMS requirements. No process related to the certified medical devices may be withheld from the Notified Body, including design, manufacture, purchase, inspections, etc. This applies to processes under direct control and those carried out by suppliers and subcontractors
- The full product specifications, including qualitative and quantitative descriptions of the product composition and list of components. No product specifications of the certified medical devices may be withheld from the Notified Body. This applies to components manufactured in-house and those purchased from external suppliers
- Initiate and maintain a systematic procedure to review experience gained from devices in the post-production phase, including the provisions referred to in Chapter VII Section 2 Article 87 of the MDR (EU) 2017/745, and to implement appropriate means to apply any necessary corrective action
- Recognize your obligation to notify the competent authorities and SGS of the following incidents immediately upon learning of them:
- Any serious incident involving devices made available on the European Union market, except expected side-effects that are clearly documented in the product information and quantified in the technical documentation, and are subject to trend reporting pursuant to Article 88
- Any field safety corrective action concerning devices made available on the EU market. This includes any field safety corrective action undertaken in a third country, in relation to a device legally available on the EU market, if the field safety corrective action is not limited to the device made available in the third country. The reports referred to in the first subparagraph shall be submitted through the electronic system referred to in Article 92 of the MDR
Pre-application review
We will review your completed and signed Medical Device Questionnaire and Product Information Questionnaire(s) and will:
- Review the completeness of the application concerning the requirements of the relevant conformity assessment procedure, as referred to in the corresponding Annex in MDR, under which approval has been sought (Annex IX or XI Part A for SGS NB 1639)
- Request additional information, if necessary
- Review the verification of the qualification of products covered by the application as devices and their respective classifications
- Review whether the conformity assessment procedures chosen apply to the device in question under the MDR
- Confirm that the devices and the conformity assessment procedures chosen lie within the scope of SGS NB 1639 designation
- Confirm that SGS NB 1639 has sufficient and appropriate resources to carry out the conformity assessment promptly
Based on the pre-application review, we will create a contract proposal. A commercial proposal and associated master service agreement will be shared with you. At this stage, we consider your application officially lodged. At this point, you can decide whether to sign the contract proposal and proceed with the certification process with SGS.
In exceptional circumstances, we may refuse the application following the pre-application review. This could be caused by an incomplete application or problems in the application documents when some devices included are outside of the SGS NB 1639 designation scope, or where we do not have sufficient resources to serve you.
Please note that any refusal or withdrawal after your application is considered lodged by SGS or later, such as after contract signing or application review (Stage 1 audit), will be notified in EUDAMED by SGS.
Contract proposal
The contract proposal, consisting of a Master Service Agreement and commercial proposal (including quotation) is submitted to you by your local SGS Delivering Office for your consideration. If the contract proposal does not adequately include all your requirements or you have questions, please contact your local SGS Delivering Office to discuss any queries and the next steps.
The contract proposal is valid for 60 days. Once the 60 days end, we will review the contract and issue a new quote, if necessary. We can only enter a contract with the legal manufacturer.
To apply for certification and to start the assessment process, the contract proposal (commercial proposal and associated Master Service Agreement) must be completed, signed and returned to us via your local SGS Delivering Office. We recommend that this is done as soon as your decision to proceed has been made, to allow maximum time for planning. Once you have signed the contract, you must notify us of any change for approval before implementation as per the dedicated section on notification of changes below.
Your responsibilities and duties as an applicant (or certified client)
The applicant retains full product liability for registered products or services, and full responsibility for correct categorization, classification and adherence to standards.
The applicant undertakes that no other application to a different Notified Body for this scope is outstanding. The circumstances of any previous Notified Body application will be documented by the applicant and sent to us before an application is accepted.
The applicant undertakes to carry out all obligations arising from a certified quality control system and applicable regulations, and maintain its adequacy and efficiency.
The application is valid for up to one year maximum after the effective date of the contract proposal (the signature date taken into consideration is the date of the signature by SGS). If the assessment has not been scheduled after this period, the contract proposal becomes void, and the applicant needs to reconfirm all submitted information to get a new contract proposal.
The applicant undertakes to inform us before implementation of any change that could impact the compliance of the device with the MDR (EU) 2017/745, affect the risk-benefit ratio or impact the clinical evaluation of the device.
The applicant undertakes to institute and maintain a post-market surveillance system according to MDR Chapter VII and to inform SGS NB 1639 in writing of any substantiated Vigilance Reports on certificated devices.
The applicant undertakes only to affix the CE Mark when all requirements of the MDR (EU) 2017/745 are met. For Class III and implantable Class IIb3 devices, this includes a valid EU Technical Documentation Assessment certificate.
The applicant is responsible for all fees and costs associated with any activity that we consider necessary to grant or maintain certification, or which are required by a European competent authority. If the proposal includes device certification with technical documentation under specific additional procedures required by MDR (EU) 2017/745 Section 5, and external scientific opinion has to be requested by the Notified Body to complete certification, associated fees, not depending on the Notified Body, will be invoiced additionally.
The applicant is responsible for informing us of all information necessary to ensure that audits, unannounced audits, assessments and communications can be efficiently and effectively undertaken, and that certification accurately reflects the current activities and product ranges, and that SGS is aware of all significant proposed changes. The changes section below gives more information.
The applicant is responsible for the right of access of us to each of its sites covered by the certification scope, including defined suppliers and subcontractors, both for unannounced audits and scheduled audits (initial, surveillance and recertification). Your contracts with relevant suppliers and subcontractors must include this stipulation. The applicant must inform us annually of any periods during which unannounced audits cannot be conducted for each of its relevant suppliers and subcontractors.
Details of the applicant’s processes for certification and control of outsourced activities are not assessed at the contract proposal stage. Therefore, if certification and control of relevant subcontractors and suppliers are found to be inadequate after application, further audits may be required, incurring additional costs.
The applicant will facilitate, as far as legally possible, the obtaining of visas for auditors to undertake audits.
The applicant takes full responsibility for the safety and security of the audit team while on-site and for scheduled audits, including advising on safe travel and accommodation arrangements, when necessary.
SGS responsibilities and duties as Notified Body (NB 1639)
We will not disclose any client information to third parties, except regulatory or enforcement authorities, where they are entitled to be informed under the MDR (EU) 2017/745. This excludes information made public in EUDAMED according to the MDR (EU) 2017/745, as this information cannot be considered confidential.
Competent authorities, including EU experts and the Joint Assessment Team, may access all information gathered during the applicant’s assessment to verify that it has been conducted by us according to MDR requirements.
We retain the absolute right to suspend, withdraw or amend the scope of certification by informing the organization and providing the reasons in writing. This includes suspension following a refusal to accept a scheduled or unannounced audit, or following undue restrictions or pressure during the audit, either at your site or that of a listed relevant supplier or subcontractor.
We retain the right to take photographs of devices and manufacturing sites, collect samples from the audit site, secure copies of documents and electronic data, and to purchase samples of devices.
We retain the right to undertake any audit, assessment or regulatory action deemed necessary to grant or maintain certification or check compliance, including visits to suppliers, subcontractors and distributors, and testing of product(s). Such activities may be carried out by us without a further application process and will be chargeable to the client. We will provide, upon request, a written explanation for the need for any additional audit, assessment, test or regulatory action. We are not obliged to inform the client before such action is undertaken.
When requested, we will provide documentary proof of the identity of unannounced audit team members and a telephone number for clients to confirm the authenticity of the unannounced audit team.
Unless stated in the proposal, it has been assumed that no further audits of suppliers, subcontractors or additional sites are required. However, during the audit process, if further information indicates a different situation, you will be informed, and additional visits agreed upon at an additional cost.
Based on the provided information, we define the on-site audit duration and sampling plan of your Technical Documentation Assessment and, after, will set up a commercial contract that would contain the price for your certification cycle and regulatory contract.
Application review: Stage 1 audit
This section does not apply to assessments conducted according to MDR Article 117.
The Stage 1 audit is conducted on-site by default, but can be conducted off-site if specific circumstances are met. The Stage 1 audit includes an appraisal of your QMS documentation and intended scope of certification, including products, processes, locations and related statutory and regulatory aspects.
This stage will include:
-
Review of all documents and elements listed in Annex IX Section 2.1:
- A draft of an EU declaration of conformity according to MDR Article 19 and Annex IV for the device model(s) covered by the conformity assessment procedure
- The documentation on the manufacturer's QMS
- A documented description of the procedures in place to fulfill the obligations arising from the QMS and required under MDR, and the undertaking by the manufacturer in question to apply those procedures
- A description of the procedures in place to ensure that the QMS remains adequate and effective, and the undertaking by the manufacturer to apply those procedures
- The documentation on the manufacturer's post-market surveillance system and, where applicable, the Post-Market Clinical Follow-Up (hereafter PMCF) plan, and the procedures put in place to ensure compliance with the obligations resulting from the provisions on vigilance set out in MDR Articles 87 to 92
- A description of the procedures in place to ensure the post-market surveillance system and, where applicable, the PMCF plan are up to date, and the procedures ensuring compliance with the obligations resulting from the provisions on vigilance set out in MDR 2017/745 Articles 87 to 92, as well as the undertaking by the manufacturer to apply those procedures
- Documentation concerning the clinical evaluation plan
- A description of the procedures in place to keep up to date the clinical evaluation plan, considering the state-of-the-art aspect
- Your quality manual, procedures and any work instructions that ensure compliance with the MDR (EU) 2017/745, appropriate common specifications and the harmonized standard for QMSs (including sterilization and other critical processes). These should be controlled and sent to the assessment team in an electronic format
- A copy of the current internal audit schedule, the last internal audit report and the minutes of the last management review to demonstrate that your internal audit and management review processes are functioning
- A list of your sets of technical documentation for the devices you wish to CE Mark, as you may be requested to send a copy of selected technical documentation to the SGS Delivering Office before the audit
- An evaluation of your location and site-specific conditions, and discussions with you to determine your preparedness for the Stage 2 audit
- A review of your status and understanding regarding the requirements of the standard(s) and regulations, concerning the identification of key performance or significant aspects, processes, objectives and operation of the management system
- A review to ensure that internal audits and management reviews are being planned and performed, and that the level of implementation of the management system confirms that you are ready for the Stage 2 audit
- Determining compliance with the documentation requirements of the MDR (EU) 2017/745 and the allocation of resources, evaluation of codes and working documentation for the Stage 2 audit
- The technical documentation is checked for preparedness to be sure it is up to date for Technical Documentation Assessment. The technical documentation itself will be assessed off-site as per the section Technical Documentation Assessment
You will receive a Stage 1/application review audit report, outlining any deficiencies (findings) to enable immediate action to be taken before moving forward. Serious deficiencies detected within the QMS during the Stage 1 audit, technical documentation preparedness, existing certification or certification of a relevant subcontractor and/or supplier could result in you being advised of additional costs and/or delays to the Stage 2 audit or Technical Documentation Assessment. A Stage 2 audit plan will be forwarded to you after the Stage 1 audit.
Stage 2 audit
This section does not apply to assessments conducted according to MDR Article 117.
This step is usually conducted several weeks after the Stage 1 audit to ensure that you have sufficient time to implement the Stage 1 audit findings. We are led by you regarding the time between Stage 1 and Stage 2 activities, but four weeks minimum would be recommended, and both stages should be planned well in advance.
A Stage 2 audit is performed on-site or as a hybrid audit (partially on-site and partially remote) and determines compliance against your documented QMS under the MDR (EU) 2017/745. This audit will also confirm the status of relevant suppliers and subcontractors, your critical processes and the eligibility of your products for medical device certification.
All assessment conclusions are based on a sampling of audit evidence to demonstrate effective implementation of the management system, control over the processes and progress made toward achieving your stated quality objectives and compliance with the MDR (EU) 2017/745.
Following the audit, the audit team will make a recommendation, dependent on the findings and the submission of corrective action plans for any nonconformances (see section on Non-Conformance and Corrective Action Requests). The auditor will talk you through the findings that may comprise major and minor nonconformances. The auditor will also agree with you on the name, address and proposed scope details that will appear on your certificates.
Technical documentation assessment
This section does not apply to assessments conducted according to MDR Article 16.
General description
The assessment of your medical device technical documentation is done in parallel to the on-site audit and is performed on a sampling basis4 for Class IIa and Class IIb. Class III, implantable Class IIb5 and Class IIb active devices intended to administer and/or remove a medicinal product are not subject to sampling, and the technical documentation of each product must be assessed.
What you need to send us:
- A complete copy of your technical documentation. Technical documentation should be submitted in English and electronically through a secured web-based application with prior agreement from SGS. This includes the part of your QMS required to support your technical documentation. All documentation should be presented in text-searchable format (i.e. text recognition PDF or Microsoft Word) and appropriately indexed to allow easy access to the relevant information. The MDR Technical Documentation Request Form can be downloaded from our website and presents a proposal of expected content for your technical documentation. It shall be completed and sent with any technical documentation
- If any relevant processes are subcontracted or outsourced, copies of any current subcontractor/supplier certification should also be sent
If the assessment of your technical documentation is leading to a high number of nonconformances that are not closed at the first follow-up review (at least 50% of the issues remain open, with consideration of their nature and severity), then we may reject the technical documentation and ask you to provide fully updated technical documentation to restart the assessment. The assessment will be charged, even if it is stopped early as the technical documentation is not compliant.
Specific procedure
Applicable specific procedures (these procedures can be subject to significant delay as external bodies are not under the jurisdiction of SGS):
- Class III implantable devices and Class IIb active devices intended to administer and/or remove a medicinal product (Rule 12 of MDR): if the device is subject to the clinical evaluation consultancy procedure as per MDR Article 54, and does not fulfill the exemption criteria as listed under Article 54(2), we will need to follow the Article 54 requirement and submit the clinical evaluation assessment report with other related documents for the CECP procedure
- Devices incorporating a medicinal substance: we shall verify the usefulness of the substance as part of the device and get a scientific opinion of the appropriate competent authority (designated according to Directive 2001/83/EC) or EMA. The manufacturer will be expected to supply technical documentation related to the drug substance, as well as the technical documentation for the device. This will be requested when we are in a position to create a usefulness report for submission
- Devices incorporating tissues or cells of animal origin or their derivatives: only devices that can claim specific additional benefits from using TSE risk species will be certified. These claims and their justification must be fully documented in the technical documentation. We shall document a summary evaluation report according to Annex II of Regulation (EU) n°722/2012 and send it to the appropriate competent authority for comments
It is a legal requirement to send any requested report to comply with the described specific procedure from MDR (EU) 2017/745 Annex IX Section 5 to the appropriate competent authorities. We will consider your comments on the established report before the consultation process starts, and begin the consultation process when your technical documentation is at the appropriate level (no nonconformances related to the specific procedure are open).
The consultation process will follow timelines set in MDR (EU) 2017/745 Annex IX Section 5. These timelines cannot be guaranteed by SGS, as the schedule for these external reviews is set by the external agency and is not under our control.
If negative feedback from the EU regulatory bodies is received, this needs to be addressed by further justification or documentation provided by you. If concerns cannot be adequately addressed, certification will not be in your interest and will not be issued despite the earlier preliminary recommendation of the reviewer.
Feedback received from the concerned competent authority will be considered for the certification decision, and the final report will be updated with the details of the external review and any actions required post-certification that are normally raised as minor CARs or as interim review requirements. The report will fully describe the device, outline your important documentation, review the history since original certification in the case of certificate renewals, and describe any outstanding non-critical nonconformances for which minor nonconformances (Corrective Action Requests) are raised. Non-critical nonconformances must be corrected within defined timescales, but do not delay certification.
After our review, the final report will either be uploaded to the European database on medical devices (Article 33, MDR (EU) 2017/745) (in this instance there will be no communication from SGS) or we will inform you of further requested action. This is part of the Technical Documentation Assessment process and will be invoiced at the same time as the device certification.
Nonconformance and corrective actions request
The Stage 2 audit, Technical Documentation Assessment and any further surveillance activities, such as a regular on-site surveillance audit, unannounced audit or device testing may lead to the identification of major or minor nonconformances by SGS auditors or products assessors.
Additional time to review and close the nonconformances will be invoiced in addition to the defined initial duration of the audit or Technical Documentation Assessment. Certification will be deferred until all major nonconformances are closed.
For further information on the closure of minor and major CARs and associated timelines, please refer to Annex 2.
Certification review
At the end of the Stage 2 and Technical Documentation Assessment, a report is compiled off-site and reviewed with other audit documentation, root-cause analysis, corrective action plans and any corrective actions taken, and a certification decision is made. This step can sometimes lead to limited changes in scope about which you will be informed. Following the outcome of the conformity assessment and associated certification decision, if applicable, the issued certificate is processed and registered in EUDAMED by SGS NB 1639.
You must be informed that certificate validity may be reduced to four years or less during the certification decision process, based on multiple aspects that would be justified to you, if relevant.
Surveillance visits after CE certification
Once issued, certificates are only valid if subject to regular audits to check the satisfactory maintenance of your QMS. Ongoing scheduled audits (surveillance visits) must be conducted annually to verify the continued implementation of your QMS according to planned arrangements, the requirements of the standard(s) and regulations.
The first surveillance audit should be scheduled within 12 months following the certification decision date. Subsequent surveillance audits must be completed within 12 months of the previous surveillance audit. Certain mandatory elements (including device testing and Technical Documentation Assessment based on sampling) will be reviewed during every visit alongside other pre-selected processes. The Medical Devices Pre-Audit Questionnaire will be sent to you before every scheduled audit, which will remind you to check recent and gradual changes. The Pre-Audit Questionnaire must be completed by you and returned to your local SGS Delivering Office well before the audit, but please remember that it does not replace the Medical Device Notification of Changes or Regulatory Action reporting obligation (see the section on Notification of Changes below).
During the surveillance audit, one or more devices will be tested (witnessing test) according to the defined sampling plan. However, if this cannot be achieved on-site, devices will be sampled and tested outside the manufacturer’s site and the fee will be added in addition to the audit cost.
Surveillance activities also cover the assessment of technical documentation, based on the established sampling plan. When requested by your local SGS Delivering Office, you must submit the required technical documentation, similar to initial certification, within four weeks of the demand to allow Technical Documentation Assessment.
An audit plan will be forwarded ahead of the agreed surveillance audit date. Please note that flexibility in the timing of ongoing visits is strictly limited by requirements defined on surveillance and renewal, as per Annex IX section 3.3, Annex VII Section 4.10 and 4.11.
Unannounced audit after CE certification
Unannounced audits can be undertaken at any time within the certification cycle, excluding prior agreed periods of unavailability. The unannounced audit cycle is associated with your certificate, so if you have multiple conformity assessment procedures leading to multiple certificates, you will have one unannounced audit cycle per certificate.
Your periods of unavailability and the ones from your relevant subcontractors and suppliers must be sent to your local SGS Delivering Office for the upcoming year, and not later than the end of each calendar year using the MDR Unannounced Audit Questionnaire. In the absence of this questionnaire, we will consider that there are no periods of unavailability. No notice will be given, so you must always be ready to facilitate these audits. Unannounced audits to investigate product compliance may be undertaken by us at any defined locations other than your site. You must help define these locations and facilitate these audits. If the unannounced audit cannot be performed, this could lead to suspension of your certificate.
Unannounced audits will focus on checking the production and traceability aspects of one of the more recent batches of devices, witnessing the final testing and inspecting processes, and auditing two processes critical to the safety and regulatory compliance of the devices. Samples may be taken for subsequent testing. You must make the technical documentation available at the audit site so it can be compared with actual or recent production.
The frequency of unannounced audits will be once every five years. However, the frequency can be increased at our discretion, following information received during audits or from other sources that devices may be nonconforming. The minimum duration of an unannounced audit is one day for two auditors simultaneously.
Recertification
As part of this program, it is not necessary to conduct a new full Stage 1 audit (application review) and Stage 2 audit. Instead, we conduct a recertification audit, (which is more in-depth than a surveillance visit). This may include an off-site document assessment and will ensure that we review all aspects of your system and technical documentation.
In the year before the expiry of your EU QMS certificate, you will be contacted by your local SGS Delivering Office to confirm your willingness to continue certification and a new contract proposal will be created. The Medical Devices Pre-Audit Questionnaire will be sent to you before the scheduled recertification audit, which will remind you to check recent and gradual changes. This Pre-Audit Questionnaire must be completed by you and returned to your local SGS Delivering Office well before the audit. Please remember that this does not replace the Medical Device Notification of Changes or Regulatory Action reporting obligation (see the section on Notification of Changes below).
In the year before the expiry of your EU Technical Documentation Assessment certificate, you will be contacted by your local SGS Delivering Office to confirm your willingness to continue certification and a new contract proposal will be created. Recertification of Class III and implantable Class IIb devices focuses on the assessment of changes, post-market activities and new risks.
For recertification of the EU Technical Documentation Assessment certificate, you will be required to complete a Medical Devices Pre-Audit Questionnaire, and provide a copy of the full technical documentation and a summary of changes and scientific findings, including all points as per Annex VII Section 4.11 Paragraph 2:
- All changes to the originally approved device, including changes not yet notified
- Experience gained from post-market surveillance
- Experience gained from risk management
- Experience in updating the proof of compliance with the general safety and performance requirements set out in Annex I
- Experience from reviews of the clinical evaluation, including the results of any clinical investigations and PMCF
- Changes to the requirements, components of the device or the scientific or regulatory environment
- Changes applied or new harmonized standards, CS or equivalent documents
- Changes in medical, scientific and technical knowledge, such as:
- New treatments
- Changes in test methods
- New scientific findings on materials and components, including findings on their biocompatibility
- Experience from studies on comparable devices
- Data from registers and registries
- Experience from clinical investigations with comparable devices
The recertification audit must be carried out and major nonconformances closed before the expiry of your current certificate.
Notification of changes
You shall inform us before implementation of any changes (see Annex 1) or regulatory actions that may affect:
- The QMS
- The scope of certification
- The device range covered
- General operations of your company
This should be reported using the Medical Device Notification of Changes, Regulatory Action, Consultancy or Services Rendered form on our website.
Planned changes cannot be implemented and devices cannot be placed on the market until formal approval of the change is issued by us in writing. If changes are implemented affecting certified devices without our formal approval, these devices would no longer be deemed covered by the issued certificate and, therefore, not legally placed on the European Union market.
We shall assess the proposed changes, determine the potential need for an additional on-site audit or Technical Documentation Assessment, and verify whether, after those changes, the QMS still meets the requirements referred to in Section 2.2 of Annex IX of MDR.
We shall then notify the manufacturer of our decision through the submitted Notification of Change form, which will be delivered to you by your local SGS Delivering Office. This will contain the conclusions of the assessment and, where applicable, conclusions of any additional on-site audit or Technical Documentation Assessment. The approval of any substantial change to the QMS or the device range covered shall take the form of a supplement to the EU QMS certificate or a supplement to the EU Technical Documentation Assessment certificate, if relevant.
The scheduling of any extension to the scope of certification can take place at the same time as the surveillance audit or recertification audit, or can be carried out between visits depending on the nature and timing of the change. This can be carried out as an on-site audit or, in some cases, by an off-site Technical Documentation Assessment. The appropriate method will be shown in the approved notification of change form and associated contract proposal.
Vigilance
Vigilance events must be reported to the relevant competent authority by the manufacturer or, for manufacturers based outside the EU, their EU Authorized Representative. Starting six months after the EUDAMED “vigilance and post-market surveillance” module is released, it must be used for vigilance reports. Until this point, it should be done via a Manufacturer Incident Form, available from the European Commission.
A copy of the report submitted to the competent authority must also be sent to us. This allows us to decide if particular actions need to be taken (such as extraordinary surveillance measures and the review of specific products or processes during the next audit) and to estimate the impact on the validity of existing certificates. It is not the role or responsibility of Notified Bodies to actively follow up on each incident so, in some cases, we will review your reports but not necessarily contact you.
Documents that must be sent are:
- Manufacturer’s Incident Report (initial, final and combined, not follow-up reports)
- Manufacturer’s Field Safety Corrective Action Report with attachments (e.g. copy of a Field Safety Notice)
- Manufacturer’s Periodic Summary Report (PSR)
- Manufacturer’s Trend Report
If you are reporting more than 10 serious incidents per month, we strongly recommend that you inform us of these in batches, in the form of a monthly report, to avoid additional administrative overheads and associated costs. Irrespective of this, serious incidents must be reported to the relevant competent authority within 15 days of you becoming aware of them or sooner, as per Article 87 of the MDR.
After our review, either the information as input for the audit team at the next scheduled audit will be filed (in this instance there will be no communication from us) or additional actions will be undertaken that must be executed as soon as possible. This could include:
- Additional information to be provided to SGS
- SGS assessing technical documentation, or specific documentation relating to the vigilance action
- SGS carrying out unannounced audit
Work undertaken by SGS as a response to vigilance reports will be invoiced.
Summary of safety and clinical performance (SSCP)
It is a requirement of the MDR (EU) 2017/745 for implantable devices and Class III medical device manufacturers to draft a Summary of Safety and Clinical Performance (hereafter SSCP) as part of their technical documentation. This summary will be validated by us and uploaded to the European database on medical devices (Article 33, Medical Device Regulation (EU) 2017/745).
An SSCP must be submitted as part of technical documentation or a stand-alone document, based on the template provided in MDCG 2019-9. Depending on the device classification. We will validate the SSCP, either as part of the initial Technical Documentation Assessment or during the certification cycle. After this review, we will only contact you if any further action is required relating to the SSCP or, otherwise, we will upload it to the European database on medical devices.
Note that a review of your SSCP may be followed by rounds of nonconformances and a review of responses until all nonconformances are closed. The costs quoted in the proposal assume no nonconformances will be raised – the review of responses to nonconformances will be invoiced.
Periodic safety update report (PSUR)
It is a requirement of the MDR (EU) 2017/745 (MDR) for Class II (Class IIa and IIb) and Class III medical device manufacturers to:
- Prepare the Periodic Safety Update Report (PSUR) as part of their post-market surveillance activities
- Update the PSUR at least annually for Class IIb and Class III devices, and at least every two years for Class IIa devices
- Upload it annually into the electronic system on vigilance and post-market surveillance (Article 92 of MDR) for Class IIb implantable and Class III only, and biennially for Class IIa implantable devices
- Make PSURs available to the Notified Body involved in the conformity assessment and, upon request, to competent authorities for devices other than Class III or implantable devices
We will assess the PSUR of Class III devices or implantable devices. We will upload its evaluation report, detailing any actions taken, in the electronic system on vigilance and post-market surveillance (Article 92 of MDR).
After the assessment of PSUR, we can undertake additional actions, including:
- The provision of additional information to SGS
- SGS assessing the technical documentation for the device, or specific documentation relating to the PSUR update
- SGS carrying out an unannounced audit
Work undertaken by us relating to PSUR updates will be invoiced.
Manufacturers of Class I devices shall prepare a post-market surveillance report and update it when relevant. This report may be requested by the competent authority.
This activity is undertaken off-site and will be identified in your proposal as an activity. If there is any appropriate action to be taken resulting from the PSUR assessment, you will be notified by us in writing.
Voluntary change of notified body
If you hold certificates with another Notified Body, you may decide to undertake a voluntary change of Notified Body to us at any point in the certification cycle (MDR Article 58). A voluntary change of Notified Body can only take place while your current certificates are valid. If you are uncertain whether you meet the criteria, contact us to discuss your options for certification with SGS.
We will review your existing certification and provide a proposal to either take over this certification within the existing cycle or start a new cycle, based on our risk assessment. In case the certification cycle is transferred, the first SGS audit should take place according to the existing audit schedule.
To initiate your voluntary change of Notified Body to SGS you need to apply as explained in the section above (Lodging your application). In addition, the following documents must be submitted:
- Copies of audit reports from your current Notified Body certification cycle
- Copies of nonconformances raised in your current certification cycle
- Copies of Technical Documentation Assessment Reports (if applicable)
- Copies of labeling (including, e.g. labels, Instructions for Use) for MDR Class III, implantable Class IIb6 and Class IIb active devices intended to administer and/or remove a medicinal product
- A summary of complaints since your last audit
- A summary of any incident reports or regulatory actions since your last audit
- Date of validity of your current certificate
- Additionally, you may submit, or we may request additional information, such as:
- The last batch or series number under your current Notified Body responsibility
- The PSUR for your Class IIa, Class IIb and/or Class III devices
- The SSCP report for MDR Class III and implantable Class IIb and Class IIb active devices intended to administer and/or remove a medicinal product
In addition to the normal contract proposal, we will send you a Medical Device Voluntary Change of Notified Body declaration to complete and sign.
If the certification to be transferred is not current or valid at the time of voluntary change, we cannot issue valid certification by the voluntary change process and any certification may be subsequently withdrawn.
During the voluntary change process, from contract signature to certificate delivery, we may contact the current Notified Body to reconfirm the validity of the certificates being transferred, and agree on the transfer date – the date when the existing Notified Body certificates will be withdrawn and SGS certificates issued. Under normal circumstances, you will be covered by a valid certificate throughout the transfer process and your ability to place devices on the European Union market should not be disrupted.
The application for a voluntary change of Notified Body remains valid for up to one year maximum after the effective date of the contract. If the voluntary change assessment has still not been planned after one year, the contract proposal becomes void. The applicant would then need to sign a new contract with SGS to continue the process.
After the transfer of Notified Body is completed, the applicant must update all labeling and other references to the previous Notified Body to refer to Notified Body 1639 and SGS Belgium N.V., as appropriate. This must be done as soon as possible, and no later than six months after the transfer date.
Application review: Stage 1 audit
The first part of the application review will be a Stage 1 audit, which follows the process described in the “Contract Proposal” section above.
Transfer audit
Following the Stage 1 audit, a second on-site review is carried out. The checks made and documentation reviewed at this stage will allow us to complete the voluntary change of Notified Body. The duration of the audit will be detailed in the contract proposal and is generally equivalent to a Stage 2 audit, as described in the “Contract Proposal” section above.
All existing nonconformances issued during the current certification cycle will be reviewed, and any nonconformances for which an effective corrective action is not demonstrated will be raised by us as corrective action requests.
If your certification covers Class III, implantable7 Class IIb or Class IIb active devices intended to administer and/or remove a medicinal product, we will carry out an additional off-site review of the submitted documentation to verify device conformity.
The audit team will issue a recommendation based on the audit findings. The auditors will also agree with you the name, address and scope details to be included on your certificate(s), where these may differ from the existing certificates.
Serious deficiencies with the QMS and technical documentation, preparedness, existing certification or certification of a relevant subcontractor and/or supplier could result in you being advised of additional costs and/or delay to the Stage 2 audit or initial assessment of the technical documentation.
Other sections of this document apply.
Additional SGS medical device certification services
The global regulatory landscape for medical device products and services is complex and many of our clients have a global reach. We provide a broad portfolio of certification and accreditation services, covering various national and international requirements. Whether your organization currently has a global reach or plans to enter additional markets, we can support your certification journey with a service tailored to your needs.
We currently provide certification services including:
- ISO 13485 (medical devices QMS)
- The MDSAP Program
- UKCA (UK MDR)
Useful references
- The European Commission portal on the MDR provides a broad range of up-to-date information and guidance documents, including:
- Common Specifications provided by the Medical Device Coordination group, which represent a set of technical and/or clinical requirements other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system
- Guidance published by the Medical Device Coordination Group, which is considered to represent a consensus interpretation of the regulatory requirements and is considered by Notified Bodies as part of their conformity assessment
- European Harmonised Standards, while not strictly mandatory, are applied by most manufacturers to demonstrate MDR compliance and, therefore, are recommended. The list of applicable standards is available on the Commission website.
- It is highly recommended that you apply the standards EN ISO 13485 and EN ISO 14971 when constructing your QMS and technical documentation
For more information on our services, visit our Medical Devices Regulatory Compliance page.
Annex 1: Changes that must be notified to SGS before implementation
| Impacted element | Significant change that needs to be notified |
| Change to the intended purpose or indications | All changes to the intended purpose or indications |
| PMCF conditions | Any change to the agreed PMCF plan |
| Change or addition of an operative technique or manual | All changes to the operative technique or manual |
| Change to the design specification | All changes impacting design specifications |
| Change to the device performance or specification | All changes to the device’s performance or specification |
| Change of software | Any impact on diagnosis, delivered treatment or software validation |
| Product range | Adding or withdrawing devices included in an existing range |
| Material | Any replacement of material to the device or device’s component |
| Mode of operation | Any change |
| Change to the sterilization method | Any change |
| Change or new subcontractor/supplier | Any change of subcontractor/supplier |
| Manufacturing process and equipment | Any change impacting manufacturing validation |
| New cleanrooms or significant change (e.g. expansion) of existing cleanroom | Any change |
| Labeling | Any change in labeling, except correction of minor grammatical/typographic errors or aesthetic changes (e.g. change in color, updated logo) |
| Change of packaging (all levels) | Any change |
| Change in the shelf life | Any change to the device’s shelf life |
| Agreed conditions or plan for CAPA on remaining open minor CAR | Any extension in time Stop or reduction of agreed study |
| Contractual data | Company name/address/number of employees/legal entity |
| Management representative | Any change |
| The Person Responsible for Regulatory Compliance | Any change |
| Change in the QMS | Any change |
| A significant change in the number of personnel (based on IAF_MD9, Annex D, Table D.1) | Any changes affecting audit time calculation per International Accreditation Forum document IAF MD 9, Annex D, Table D.1 |
| Design/manufacturing/ labeling/ packaging change related to CAPA | All changes |
| Data on the certificate | Any information recorded in EUDAMED |
| Changes that impact compliance with GSPR (Annex I) | All changes |
| Changes that impact the device’s safety and performance | All changes |
| Incidents outside the EU triggering FSCA impacting devices sold in Europe | All changes |
| Changes that impact the device risk profile | All changes |
This is a non-exhaustive list of examples. In case of doubt, please contact your local SGS Delivering Office.
Annex 2: Corrective Action Request (CAR)
A QMS on-site audit and/or Technical Documentation Assessment may detect and record one or more nonconformances (non-fulfillment of a requirement). These nonconformances are presented to you in a Corrective Action Request (CAR) form for all QMS-related nonconformances. For Technical Documentation Assessments, nonconformances or potential nonconformance are integrated directly into the assessment report as Potential Issues to be Clarified (PIC) at the initial assessment and, if not solved, as major CARs in the follow-up report. Both forms are formal requests to describe the specific corrections and corrective actions taken, or planned to be taken, to eliminate the detected nonconformances within a defined timeframe. In addition, for QMS nonconformances on-site, you are requested to analyze the root cause of the nonconformances and provide us with corrections and corrective actions.
Please be informed that any identified major CARs can result in a recommendation of suspension of your device or certificate by the SGS Audit Team Leader or Product Assessor. Therefore, every major CAR shall be given the appropriate consideration for review and closing.
We would like to remind you that any delay in submitting a corrective action plan and implementing corrections or corrective actions for major CARs may lead to new certificates not being issued and current certificates being suspended, or a device being removed from the certification scope.
This document explains the underlying SGS NB 1639 process on corrective action requests, which starts from the moment of presenting the detected nonconformances to you (by the auditor and/or the product assessor). By default, the date of the nonconformance is the last day of the audit or Technical Documentation Assessment. It is very important to respect the timelines associated with CAR closure as no concession will be given.
These timelines for CAR closure are related to the severity and/or (potential) impact of the associated nonconformances and are defined by the auditor and/or product assessor according to SGS internal procedures. These timeframes are recorded and monitored by us, as well as by accrediting bodies and competent authorities.
CAR – general information
- A nonconformance can be graded as minor or major by the auditor or product assessor, depending on its severity and impact on the product’s safety.
- Make sure you understand the non-fulfillment of a requirement when the CAR is recorded.
- For each QMS CAR, an action plan is requested. Your action plan must contain a sufficient level of detail to demonstrate to the auditor that you understand the essential details of the findings of nonconformance, and that you have identified root causes and, subsequently, the corrective actions needed. If appropriate, you also need to demonstrate corrections, or you must have a sound justification for not having finalized corrections.
- The timelines associated with CAR closure are presented below:
- A specific date is scheduled and reserved for evaluating action plans and evidence, as a response to the CAR to be submitted by a client. It is vital that action plans and associated data are correct, complete and submitted on time, sufficiently resolving the nonconformance(s). It is neither SGS nor the auditor and/or product assessor’s responsibility to send reminders to ensure this information is submitted on time according to planned arrangements.
- Poor “quality” of corrective action plans and/or failure to submit on time will cause serious delays that are the manufacturer’s sole responsibility. If objective evidence is correct, complete, on time and sufficiently demonstrates the resolution of the nonconformance, the auditor and/or product assessor can close the CAR.
- If, after an initial assessment of your technical documentation, a large number of PIC have been raised, we can allow the product assessor to recommend voiding the Technical Documentation Assessment if less than 50% of the PIC (with consideration of their nature and severity) can be closed at the first follow-up review. You will be notified of this in writing in the Technical Documentation Assessment report.
| Stage | Classification | Timelines and a round of reviews |
| CARs raised at initial MDR Technical Documentation Assessment (before CE certification) | Major CARs | One round of PIC must be answered within two months, followed by a maximum of three rounds of major CAR reviews. Major CARs must be closed within one year of the date of the initial Technical Documentation Assessment report being sent to you. |
| Minor CARs (once all major CARs are closed) | A maximum of 12 months, starting from the date the minor CAR was raised and a maximum of two rounds of reviews are allowed. | |
| Minor QMS CARs (once all major CARs are closed) | Minor QMS CARs are reviewed during the next on-site audit. | |
| CARs raised at MDR Stage 2 audit (before CE certification) | Major CARs | Two rounds of major CAR reviews (major CARs must be closed within one year of the last day of the Stage 2 audit). |
| Minor CARs | Minor QMS CARs are reviewed during the next on-site audit. | |
| CARs raised at MDR Surveillance Technical Documentation Assessment (after CE certification) | Major CARs | One round of PIC must be answered within two months, followed by a maximum of two rounds of major CAR reviews. Major CARs must be closed within six months of the date of the initial Technical Documentation Assessment report being sent to you. |
| Minor CARs (once all major CARs are closed) | A maximum of 12 months, starting from the date the minor CAR was raised and a maximum of two rounds of reviews are allowed. | |
| Minor QMS CARs (once all major CARs are closed) | Minor QMS CARs are reviewed during the next on-site audit. | |
| CARs raised at MDR Surveillance on-site or unannounced audit (after CE certification) | Major CARs | Major CARs should be closed within 90 days and two rounds of reviews. |
| Minor CARs | Minor QMS CARs are reviewed during the next on-site audit. |
To close a major CAR from a QMS on-site audit, the following steps must be followed:
- Conduct root-cause analysis, define the appropriate correction and set the relevant corrective action plan immediately
- Send the corrections and corrective action plan to the auditor as soon as possible, but within two working days following the receipt of the CAR
- The auditor comments on the action plan or accepts it as presented. However, it remains your sole responsibility to resolve the findings of nonconformance. The action plan is a precondition to demonstrate the appropriate intended actions to the auditor and give confidence in a successful review and closure at the planned date
- Documented evidence of the corrections and corrective actions implemented or being implemented must be sent to the auditor no later than 30 calendar days following the opening of the major CAR
- The auditor reviews the evidence and determines if it is acceptable. If the provided evidence is not acceptable, the auditor will provide their feedback in writing, including the date at which you must send corrected evidence of the corrections and corrective actions implementation
- Only two iterations of evidence sent and auditor’s feedback are authorized within one year (for the initial certification audit) or 90 days (for the surveillance or unannounced audit). If, by the second review, the provided evidence is not satisfactory, the expected new certificates will not be issued and current certificates will be suspended, or the corresponding device removed from the certificate scope. Nevertheless, the major CAR needs to be resolved, reviewed and closed after the second iteration. Unresolved CARs cannot serve a future lift of suspended certificates
- If the auditor has determined that a follow-up visit on your site shall be performed to close the major CAR, the follow-up visit must be organized after a review of evidence and within 90 days. This visit will evaluate actions taken, their effectiveness, and determine whether certification can be granted or continued
- If a major CAR is not closed as per established timeliness, certification will be at risk of suspension or withdrawal. Suspended and withdrawn CE certificates are automatically reported to the relevant competent authority and in EUDAMED
- Successful review and closure of all open CARs will lift (potential) sanctions on certification unless certificates have been withdrawn permanently
To close a major CAR resulting from Technical Documentation Assessment, the following steps must be followed:
- Send documented evidence of the corrections and corrective actions implemented or being implemented to the agreed SGS contact, no later than 30 calendar days following the opening of the PIC or major CAR
- The product assessor reviews the evidence and determines if it is acceptable. If the provided evidence is not acceptable, the product assessor provides their feedback in writing, including the date at which you must send updated evidence of the corrections and corrective actions implementation
- Only four iterations (for CARs raised during the initial Technical Documentation Assessment) or three iterations (for CARs raised during the surveillance Technical Documentation Assessment) of evidence sent, and product assessor’s feedback are authorized within the one year or six months’ timeframes, respectively. If by the second review, the provided evidence is not satisfactory, the expected new certificates will not be issued or current certificates will be suspended, or a device may be removed from the certificate scope
- To reinstate the product on the certificate or lift the suspension, the major CAR needs to be resolved, reviewed and closed
To close a minor CAR from a QMS on-site audit, the following steps must be followed:
- Conduct root-cause analysis, define the appropriate corrections and set the relevant corrective action plan immediately
- Send the correction and corrective action plan to the auditor as soon as possible, but no later than within two working days following the receipt of the CAR
- The auditor comments on the action plan or accepts it as presented. However, it remains your sole responsibility to resolve the findings of nonconformance. The action plan is a precondition to demonstrate the appropriate intended actions to the auditor and indicates a successful review and closure at the planned date
- Implement your corrections and corrective actions according to your plan and prepare documented evidence for the next SGS on-site audit
- The review will take place during the next scheduled on-site audit. Evidence of corrections, root-cause analysis and corrective actions will be reviewed. In the case of multi-site companies, where sites are sampled during the next planned audit, the review will be performed on the main site/headquarters
- Any minor CARs that cannot be closed on time will be automatically upscaled to major CARs
To close a minor CAR resulting from technical documentation assessment, the following steps must be followed:
- Send documented evidence of the corrections and corrective actions implemented or being implemented to the agreed SGS contact, as determined on the CAR form by the product assessor. This is normally within one year of the date the minor CAR was raised
- The product assessor reviews the evidence and determines if it is acceptable. If the provided evidence is not acceptable, the product assessor provides their feedback in writing, including the date at which you must send updated evidence of the corrections and corrective actions implementation
- Only two iterations of evidence sent and the product assessor’s feedback are authorized. If, by the second review, the provided evidence is not satisfactory, the minor CAR will be upscaled to a major CAR. If the client fails to address the major CAR within the defined timeline for major CAR closure, the product or certification will be at risk of withdrawal or suspension
Guidance on root-cause analysis and corrective action/preventive action (CA/PA):
- Correction:
- Root-cause analysis:
- Corrective action:
- Corrective actions always need diligent review and verification to ensure that the new situation will not introduce new causes of identical, similar or other deviations
- General:
- Preventive action:
1Except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling
2Please note that for systems and procedure packs (MDR 2017/745 Article 22), the System and Procedure Pack Product Information Questionnaire (available on the SGS website) shall be submitted instead of the Product Information Questionnaire
3Except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling
4Devices are sampled according to MDCG 2019-13
5Except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling
6Except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling
7Except for sutures, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors, which are subject to sampling